Craig’s Discussion of Study
The study below, “Bioelectric Control of Metastasis in Solid Tumors” is one of many that establishs that the immune system’s control of solid cancers is fundamentally bioelectric. I also include a discussion about canine Neosporosis – if your dog has this (mine had it), I suggest you skim through some of the first few sections to get the fundamentals for the dedicated Neosporosis section.
When I studied molecular biology and biochemistry at the University of Oregon, we were trained to think of physiology in the typical antigen/antibody or as chemical reactions. Bioelectricity was not, and still is not, getting the deserved recognition it deserves as the fundamental consideration of biological processes.
What these many studies demonstrate is the immune system’s tumor-destroying mechanisms are bioelectrically regulated by oxidation and reduction (passage of electrons). These studies have also found cancer cells having much different voltage potential metrics.
“cancer cells have a more positive resting membrane potential than other somatic cell types, in the range of −30 to −20 mV, making them more similar to proliferative cell types such as embryonic and stem cells than healthy differentiated cells.”
See below study
Simplify stated, the reactions that promote tumor destroying go in the “opposite” direction of the tumor promoting (technically, supression of tumor destroying).
Oncologists refer to a “tumor microenvironment” (TME) to understand a particular cancer’s dynamics (even for an individual). What I’m saying is, while yes the many molecular/genetic factors currently discussed about the TME, I have yet to hear analysis of the bioelectrical component, save for a few studies that boldened my hypothesis.
In these studies, researchers measured Total Antioxidant Capacity (TAOC) and not only did they find cancer patients having far less than healthy individuals, there is also eveidence that the resistance to therapy that cancers are known to develop are ALSO correlated to TAOC.
Le Chatelier’s Principle states: “When any system at equilibrium for a long period of time is subjected to a change in concentration, temperature, volume, or pressure, (1) the system changes to a new equilibrium, and (2) this change partly counteracts the applied change.”
Using this principle, if tumor generation (IMO) is the imbalance of oxidants (“reactive oxygen species”, to be technical) and antioxidants then, until we bring the TME to bioelectrical homeostasis, therapies will either be ineffective or not durable (they don’t last).
My Observations – Immuno-Oncology
I work for an immuno-oncology company, and I was recently looking at AE’s (adverse events) of patients in clinical trials and noticed the high amount of electrolytes such as potassium (+ charge), after treatments.
When a cancer therapy starts destroying a tumor, the tumor breaks down and goes into the blood stream. If this happens too fast for the body’s kidneys, it can lead to Tumor Lysis Syndrome, which can be fatal.
Immunotherapies such as Immune Checkpoint Inhibitors (ICI’s) got everyone excited in the last ten years because they made so much sense from a molecular POV, AND they demonstrated from partial to complete responses in patients. Yea!
But then we saw HIGH percentages of complete response patients have the tumors come back, often more fiercely.
Back to the potassium.
Knowing that a positively charged bioelectrical imbalance favors tumor growth, well then that suggests as tumors are eliminated by ICI’s, chemotherapy, radiation, etc., the electrolyte byproducts of the tumors might be further imbalancing the bioelectrical environment. So UNTIL we create homeostasis, this logic predicts cancer therapies to develop resistance, which is exactly what is happening.
Restoring Bioelectrical Homeostasis
Earthing
In short: ground yourself by walking barefoot or using a grounding mat. The antioxidant benefit from the earth is in an entirely different (and more effective) class than vitamins containing anti-oxidants.
HOW IT WORKS
The earth has a negative charge giving electrons to those in contact with it, and the atmosphere has a positive charge. In the early 60’s, humans started walking in shoes with rubber (insulating) heels, cutting us off from the earth’s negative charge.
Since that time, chronic inflammation took off. Inflammation is the immune response correlated to the low TAOC serum levels mentioned earlier. In other words, the shift from bioelectric equilibrium favored the inflammatory response as Le Chatalier’s Principle predicts.
The more we learn about inflammation, the more we’ve realized it is linked to everything from cancer to Alzheimer’s Disease.
If you haven’t heard of earthing, you MUST spend ten minutes watching Clint Ober’s interesting and well-produced documentary.
If you’re a scientist and want to see the evidence from peer reviewed studies, there are 791 such studies in Google results.
Cancer in Dogs
Dogs touch the earth with their paws daily, grounding them briefly. Dogs should be given grounding mats or other such grounding devices for inside the home. It is especially important to sleep grounded, as our bodies do their most productive healing and tumor monitoring and fighting during this time.
Modern society and working owners have resulted in walks outside that are typically brief and infrequent. Advanced cancers further reduce the amount of time a dog is outside, perhaps accelerating cancer growth.
Dogs with Neosporosis
This is an interesting one. My dog Oscar died of it and it was by far the most painful experience of my life – not only was he my best friend, but Neosporosis is about the cruelest way to go imaginable. I spent over 200 hours reading research coming out of labs each and every week in pursuit of a therapy more effective than the few currently available.
If your dog has Neosporosis, this is what you need to know.
Most veterinarians know little, some none, about Neosporosis and often misdiagnose. Even the “specialists” I brought Oscar to didn’t know much. In fact, veterinary neurologists treat this because it is a progressive paralysis, HOWEVER it is an infection caused by a protozean whose “cousin” causes toxoplasmosis in humans (rare in US but elsewhere as many as 2/3rd get it).
Neospora is an “intra-cellular” infection, a particulary nasty one wherein it enters a cell and hijacks organelle machinery, then reprograms them to make lipids (fats), as Neospora cannot make them themselves (probably why they target dogs – high fat diets).
The important takeaways is they are inside cells and your dog fights them with a police SWAT tactic – by essentially gassing them with nitrous oxide.
Nitric oxide is one of those immune system processes that requires a lot of oxidation and reduction, the processes that pass electrons around to regulate the immune response while maintaining homeostasis.
Your dog’s Neospora fight requires a very high Total Antioxidant Capacity. I realize now, in hindsight, that of all the dogs in the Neosporosis support group I heard about, the only one who eliminated it was being fed berries (very high antioxidant) and a special diet I can’t quite recall, AND was frequently outside.
Don’t give your dog Vit B12 shots. B12 reduces nitric oxide levels in the body. Someone in the support group announced her naturopathic vet had the bright idea of B12 injections. I pleaded and pleaded not to, telling her straight up it would kill the dog. But I’m not a vet, as the vet pointed out.
The dog was dead within two weeks.
In fact, recent studies link the nitric oxide synthase enyzme to parasite restriction in dogs with Neosporosis as a new targeted therapy.
Nitric oxide is so key to your dog’s ability to resist the infection that the first thing Neospora does upon getting into the dog is shut down Nitric Oxide synthesis in a variety of ways – including an exhaustive series of tactics designed to inhibit the synthesis of Nitric Oxide building blocks.
I won’t go into detail on that in this post (I intend to write an ebook about it – if your dog has Neosporosis and you’d like to learn more, comment below), but I developed a treatment regimen for Oscar that induced nitric oxide and dosed him with catalytic enzymes Neospora was fiercely trying to shut down.
So take away – there are tools for Neosporosis that your vet is unlikely to know anything about. Use them.
OK – the Bioelectrical Science

Abstract
As the leading cause of death in cancer, there is an urgent need to develop treatments to target the dissemination of primary tumor cells to secondary organs, known as metastasis. Bioelectric signaling has emerged in the last century as an important controller of cell growth, and with the development of current molecular tools we are now beginning to identify its role in driving cell migration and metastasis in a variety of cancer types. This review summarizes the currently available research for bioelectric signaling in solid tumor metastasis. We review the steps of metastasis and discuss how these can be controlled by bioelectric cues at the level of a cell, a population of cells, and the tissue. The role of ion channel, pump, and exchanger activity and ion flux is discussed, along with the importance of the membrane potential and the relationship between ion flux and membrane potential. We also provide an overview of the evidence for control of metastasis by external electric fields (EFs) and draw from examples in embryogenesis and regeneration to discuss the implications for endogenous EFs. By increasing our understanding of the dynamic properties of bioelectric signaling, we can develop new strategies that target metastasis to be translated into the clinic.
Metastasis is the leading cause of death in patients with solid tumors. Metastasis describes the dissemination of tumor cells from the primary tumor to secondary organs in the body via the lymphatics, vasculature, or cerebrospinal fluid. In this review, we primarily focus on solid cancers, such as prostate, breast, skin, lung, colorectal, and glioblastoma. Progression of these cancers requires cell migration out of the primary tumor into local tissues through various physical barriers, which is driven by components of the local tumor microenvironment and executed by complex signaling pathways in the cell. There is no single dominant pathway that controls metastasis, making it an extremely challenging process to study. There is a critical need to better understand the mechanisms driving metastasis to identify novel strategies to prevent, diagnose, and treat metastatic cancer.
Since Burr first reported the presence of a tumor in vivo using voltage readings in 1941,1 studies have demonstrated the role of bioelectric signaling in cancer cell proliferation and tumor growth. Here, we focus specifically on the role of bioelectricity in regulating cancer cell metastasis, reviewing the ways in which ion channel expression, membrane potential changes, and external electric fields (EFs) have been implicated in regulating invasion and metastasis. We also highlight the implications of the emerging field of developmental bioelectricity for translation of new biophysical controls of cell behavior to the clinic.
Metastasis—An Overview
Metastasis is a multistep process that involves the following events: local invasion to surrounding tissues, intravasation into the vasculature or lymphatics, survival and transit in the vessels, and extravasation and colonization in a secondary organ2,3 (Fig. 1A).

Invasion
Cancer cell invasion is the first step of metastasis, through which a cell disrupts its basement membrane and invades into the surrounding stroma. Invasion occurs due to tumor cell extrinsic changes in the microenvironment that attract tumor cells into the local tissue, and the activation of signaling pathways within tumor cells at the genetic and protein level that enable cell motility and extracellular matrix (ECM) degradation. Several cues within the tumor microenvironment can promote local invasion.4 For example, fibronectin, an ECM protein that provides structure and support to tissues, can attract breast cancer tumor cells to the vasculature via haptotaxis (i.e., directional migration in response to substrate-bound cues) to promote dissemination.5
Soluble cues such as growth factors and cytokines can also attract tumor cells via chemotaxis to promote invasion.6 Local invasion is driven by signaling pathways that promote cytoskeletal dynamics and promote cell motility, which have been extensively described in other reviews.7–11 Cells can migrate in different modes: either individually or collectively, as groups of cells held together via cell/cell interactions. Individually, cells can take on mesenchymal cell movement driven by lamellipodial extension, which requires cell-matrix or amoeboid-like movement. Here we focus on lamellipodia-based cell migration, given that all evidence for participation of electric signaling in migration is with this type of migration.
To migrate, a cell first extends actin-rich protrusions such as lamellipodia and filopodia (Fig. 1B). Then, focal adhesions will form at the leading edge, which help the cell propel itself forward, eventually retracting the trailing edge via disassembly of focal adhesion, mediated by calpains. Invadopodia are another type of invasive structure used by cancer cells to locally degrade basement membrane and promote migration. Detailed mechanisms of cell migration have also been reviewed elsewhere. The secretion of proteases by invading cells is important for local ECM degradation, enabling cells to move within the ECM. Finally, cancer cell migration can also be facilitated by the epithelial to mesenchymal transition (EMT), a developmental process driven by specific transcription factors to promote a more plastic and invasive phenotype.3
Intravasation, vessel survival, and extravasation
Intravasation describes the entry of tumor cells into the vasculature or lymphatics. Macrophages play an important role in attracting tumor cells to the blood vessels and enhancing vessel permeability to enable intravasation.10 The exact mechanism by which tumor cells enter the vasculature remains unclear. Studies suggest that tumor cells do not entirely disrupt endothelial tight junctions when they intravasate, with others positing that tumor cells can enter vessels via entosis, the invasion of one cell into another.12 Once they have entered the vasculature or lymphatics, tumor cells are referred to as circulating tumor cells (CTCs), which can be detected for all solid tumors. Here, they exist as both individual and multicellular cells with a combination of both epithelial and mesenchymal traits.13
Colonization
While hundreds of thousands of cancer cells can enter the bloodstream,14 a majority of CTCs do not successfully lead to metastasis formation within a secondary organ. For example, using a mouse model of melanoma, one study demonstrated that only 2% of injected cells will form micrometastases, with less than 0.05% forming secondary tumors.15 Once tumor cells have entered a foreign tissue, they are called disseminated tumor cells (DTCs). DTCs can remain dormant, exhibiting prolonged survival in a foreign microenvironment, reversible growth arrest, and resistance to therapies.16 The transition of a tumor cell (DTC) into an overt metastasis is highly dependent on the local microenvironment of this organ. The original “seed and soil” hypothesis by Stephen Paget suggested that tumor cell dissemination was not random, but instead confined to specific organ sites based on favorable interactions between tumor cells and the host environment.17 The formation of a supportive premetastatic niche, composed of ECM and resident immune cells, is essential to provide nutrients and survival signals that drive DTC survival and outgrowth.18
While our understanding of the mechanisms that drive the different steps of metastasis has increased, these are yet to be successfully translated into clinical applications. Currently, most metastatic carcinomas are treated with chemotherapeutic drugs, which inhibit cell proliferation and promote cell death by targeting the cell cycle or cell division. More recently, targeted therapies based on the identification of specific driver mutations, which have been facilitated by precision medicine, as well as immunotherapy, have emerged as treatment options for patients with metastatic disease. Steeg and Anderson et al. comprehensively discuss strategies for targeting metastasis.19,20 Most current clinical trials assess growth by the clinical RECIST criteria, which measure tumor size and ignore the ability of new drugs to inhibit cell motility and metastasis. Several strategies are being developed to improve patient outcomes. First, biomarker-based tests such as MammaPrint Dx for breast cancer21 and Decision-Dx-UM for uveal melanoma22 have been developed to predict the metastatic potential of a newly diagnosed tumor. Second, it is now clear that tumor cells can become invasive and disseminate early in cancer development, suggesting that targeting the premetastatic niche in secondary organs may provide an alternative approach to preventing metastatic disease.16,18 Finally, there is a renewed focus on the identification of new drugs that specifically target already disseminated disease, rather than only targeting the primary tumor.
The development of effective treatment approaches that target metastatic cancer requires an understanding and exploitation of the signals that normally keep cells operating toward the anatomical needs of tissues and organs—limited growth and tightly orchestrated morphogenesis. In complement to the focus of the field on secreted molecules, other biochemical factors, and proteolysis, here we review the state of the art with respect to an important physical signaling modality: bioelectricity,23–25 which mediates many of the normal cellular functions that go awry in metastasis.
Bioelectricity in Cancer
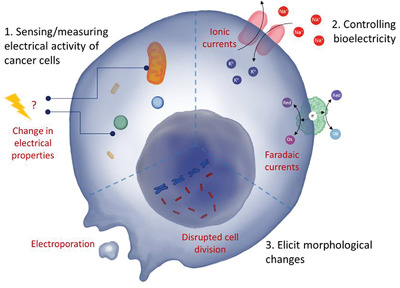
All cells possess an electric potential across their plasma membrane. Although perhaps most familiar in the context of excitable cells such as neurons, nonexcitable cells are also able to generate and receive bioelectric signals.26,27 The combined electrochemical gradients across a membrane produce the membrane potential, or Vmem. When the cytoplasm becomes more positively charged relative to the extracellular space, the cell is said to be depolarized and will have a less negative Vmem. When the cytoplasm becomes more negatively charged relative to the extracellular space, the cell is said to be hyperpolarized and will have a more negative Vmem.
Physiological Vmem can range from −90 to −10 mV, depending on the cell type and physiological state28,29 (Fig. 2A). The membrane potential is regulated by ion channel expression in cells, ionic composition of the extracellular milieu, and the presence of bioelectric gradients, such as an EF, within a tissue.30 Bioelectric signaling is recognized to regulate many processes important for cellular homeostasis by establishing biochemical gradients, altering gene expression, and modulating cell signaling. Ion and voltage gradients have been functionally implicated in the control of cell differentiation, migration, and proliferation, with important instructive roles during embryogenesis, regeneration, and remodeling in a wide range of model systems, including human embryos and cells in vitro.31–33

Although cells can express each channel type in varying numbers, it is the sum of ion flow, based on activity of the expressed channels, that will establish the overall charge of the cell (Fig. 2B). At the tissue level, the bioelectric state of one cell can also influence neighboring cells and EFs are generated by the membrane potential of a collection of cells, propagating throughout the population to form charge-specific domains (Fig. 2C). EFs are important instructional cues for cell migration and tissue organization.34,35 They are present in adult tissues such as the breast epithelium and regulate epithelial wound healing and cell migration during embryogenesis and regeneration.36–38
More than 150 years ago, Emil Du Bois-Reymond reported the presence of EFs in the skin of frog wounds,39 however, applying the principles of bioelectric signaling to the hallmarks of cancer is a nascent field of study. In 1941, Burr demonstrated that voltage readings can be used to detect the presence of a tumor in vivo, suggesting that the electrical characteristics of a tumor differ from the surrounding normal tissue.1 Thirty years later, Cone first postulated that mitosis is controlled by changes in membrane potential and ion flux, linking it to malignant states.40–42 It was not until the 1990s that momentum in this field increased and researchers began to strongly consider the bioelectric properties of cancer cells as a potential avenue for investigation. Since then, evidence has accumulated demonstrating that the bioelectric impedance of cancerous tissue differs from the surrounding tissue and that this may be a potential target for cancer therapy.43,44
Individual cancer cells have a more positive resting membrane potential than other somatic cell types, in the range of −30 to −20 mV, making them more similar to proliferative cell types such as embryonic and stem cells than healthy differentiated cells.45,46 Combined with further observations that ion channels are abnormally expressed in both cancer patient biopsy tissues and cancer cell lines, and that blocking ion channels can inhibit cancer cell proliferation, bioelectrical signaling is emerging as a hallmark of cancer.47–50 While initial studies provided evidence for the role of bioelectric signaling in cancer cell transformation and proliferation, increasingly it has become recognized that bioelectricity can also control cancer cell metastasis at both cell intrinsic and extrinsic levels, which is discussed in this review.
Intrinsic Bioelectric Properties of Cancer Cells
It has long been noted that many tumor cells have striking differences in both ion channel expression/activity and Vmem when compared with normal cells.51 These bioelectric properties include expression and activity of ion channels, and changes in Vmem. Current evidence suggests that bioelectric properties of a cell influence migration in three main ways: cytoskeleton regulation, changes in cell volume, and pH. Clearly, these properties are important for metastasis, but in many cases, it is not well known how they translate into the biochemical signaling pathways that control cell migration and invasion.
Ion channels
Ion channels are pore-forming membrane proteins that create ionic concentration gradients by regulating the flow of ions such as Na+, K+, Ca2+, and Cl−. At the basic level, they are classified into voltage gated and ligand gated. Molecular techniques have allowed for a more elaborate classification based on the subunit type and gating mechanism: physical (light, temperature, pressure, and tonicity), chemical (pH, pO2), and intracellular factors (ATP, secondary messengers). For a more detailed review of ion channel subtypes and their role in general cell migration, we refer the reader to other reviews.52–55
It is now understood that there is a host of ion channels whose expression is dysregulated in cancer cells and are associated with a metastatic phenotype. For example, microarray expression profiling of ion channel genes in patient primary tumors of breast cancer,56 lung adenocarcinoma,57 and glioma58 has identified many ion channel genes that are differentially expressed compared with normal tissues. In addition, the availability of public gene expression data sets from patient samples on websites such as cBioPortal or Oncomine, or cell lines from the Broad CCLE portal will be useful in better characterizing the ion channel expression profile of various cancers. It will be important to assess whether the ion channel expression profile of cancer cell lines is similar to that of primary tumors, and whether the primary tumor ion channel expression is similar to that of metastases.
While the expression of many ion channels is linked to a metastatic phenotype, it is the activity of the functional channels expressed that will ultimately dictate the net effect on cell migration and invasion. The main function of ion channels is to maintain cellular homeostasis by regulating the flux of ions in and out of the cell, but they are also higher order regulators of many downstream molecular signaling pathways. There are four main ions that play a role in establishing the resting Vmem: Ca2+, Na+, K+, and Cl−. Locally different ion concentrations are important for initiating cell migration,59,60 and ion channels can also work in tandem by functionally coupling to provide a positive feedback loop.61 A summary of the main contributions to metastasis by each channel type is presented in Figure 3.

Calcium
The intracellular calcium concentration (Ca2+i) is integral for cancer cell metastasis. The level of Ca2+i regulates the cell cytoskeletal dynamics, protease activity, cell volume, and pH of the cell, all of which contribute to migration and invasion of cancer cells62–65 (Fig. 3A). In many cases, the activity of other types of ion channels contributes to cancer cell migration through their indirect effect on Ca2+i. Calcium is important for cell adhesion turnover and a polarized Ca2+i concentration gradient of higher Ca2+ concentration in the leading edge of the cell and vice versa for the trailing edge is found in many migrating cell types.66–68 The protease calpain is dependent on Ca2+ and will inactivate E-cadherin to facilitate the local disassembly of focal adhesions to allow detachment of the rear part of the cell, facilitating migration.69
Calcium is also involved in promoting EMT pathways and the activity of matrix metalloproteinases (MMPs) to drive ECM degradation and cell invasion.70,71 Many Ca2+ channel and exchanger genes are overexpressed in solid tumor cancers, although it is worth noting that the Ca2+I level, dictated by channel activity, is important in determining which pathways will become activated in the cell. For example, influx of Ca2+ can promote the activation of cell cycle pathways and migration, but elevated Ca2+i can also cause cell apoptosis.72,73
Several channels from the transient receptor potential melastatin (TRPM) cation channel family have been shown to play a role in cancer metastasis. Some, such as TRPM2 and 7, have an inhibitory effect on metastasis. TRPM2 facilitates Ca2+ entry into cells experiencing oxidative stress to increase cell death, of which cancer cells have adapted mechanisms to overcome.74–76 TRPM7 responds to extracellular acidification, a common feature of the tumor microenvironment, by increasing the inwardly directed cation conductance, making the channel less selective for Ca2+. This leads to a reduced Ca2+ influx and thus reduced activation of downstream Ca2+-dependent migration pathways.77
The role of other TRP channels is less clear and may involve a metastasis-inducing function. In prostate cancer cell lines, TRPM8 stimulation inhibits migration,78 and TRPC1 and TRPM8 expression is associated with noninvasive and well-differentiated tumors in breast ductal adenocarcinoma tissue.65 However, others have reported that overexpression of TRPM8 channels in breast cancer cell lines increases metastatic potential by promoting EMT via the Akt/GSK3β pathway,79 and that TRPM8 expression in glioblastomas increases cell migration.80 This suggests that these channels may have cancer or cell type-specific functions.
Finally, TRPV2, which is normally translocated from the endoplasmic reticulum (ER) to the plasma membrane in response to PI3K-activating ligands and mechanical stress,81 is expressed at higher levels in metastatic tissue compared with the primary tumor.82 In androgen-resistant prostate cancer cells, it is translocated and constitutively activated to maintain elevated Ca2+i to both promote cell migration and the expression of MMP2 and MMP9 for ECM degradation.82,83 Furthermore, TRPV2 was reported to be recruited via PI3K/Akt signaling to the pseudopodia in three different metastatic breast cancer cell lines where it increased Ca2+i and cell migration synergistically with the Ca2+-activated K+ channel KCa1.1 to create a positive feedback loop for continued Ca2+ influx.84
Another mechanism known to influence metastasis via calcium flux is store-operated Ca2+ entry (SOCE). This involves two proteins: Orai1, a plasma membrane channel that opens in response to depleted ER Ca2+ stores, and membrane-localized stromal interaction molecule 1 (STIM1) that acts an intraluminal Ca2+ sensor and regulates the opening of SOCE channels.54 Orai1/STIM1-dependent Ca2+ influx is necessary for the turnover of focal adhesions in breast cancer cells by promoting the activity of calpain.85,86 Another Ca2+-activated K+ channel, KCa2.3 (aka SK3), couples with Orai1 in lipid rafts of breast and colon cancer cells to increase Ca2+-dependent protease calpain.62 The colocalization of Orai1 and KCa2.3 was confirmed in breast cancer patient samples and metastasizing transplanted tumors in an animal model of breast cancer.87
The last major types of ion channel involved in Ca2+-driven metastasis is L-type voltage-gated calcium channels (VGCCs) and calcium-permeating Piezo type mechanosensitive ion channel components (Piezo1 and 2). For the former, integrin activation and signaling through Src kinases stimulate VGCCs to increase Ca2+i in the filopodia tips of cancer cells. Localized increase of Ca2+ in filopodia then activates calpain to facilitate directional cell migration via filopodia stabilization and focal adhesion regulation.63 These VGCCs can be targeted with Ca2+-channel antagonists in breast cancer.88 For mechanosensitive calcium channels, it has been demonstrated in glioma and melanoma cells that Piezo1 and Piezo2 channels can regulate cell motility by sensing physical cues from the microenvironment to generate a calcium signal.89,90 In particular, Piezo1 has been shown to be localized at focal adhesions in glioma cells and regulates their assembly.91
Sodium
Although calcium is a key ion in the main cell migration signaling pathways, many studies have demonstrated that cancer cells can also effectively use Na+ flux to indirectly promote a metastatic phenotype (Fig. 3B). A change in Na+ flux can create localized areas of depolarization that drive the movement of other ions such as Ca2+ and H+. There is also a link between Na+ and Ca2+ to control the invasion machinery of the cell; mitochondria possess an Na+/Ca2+ exchanger that can take up excess Na+, resulting in an accumulation of cytosolic Ca.2+92 An increase in intracellular Ca2+ can then initiate many of the signaling cascades described in the previous calcium section, such as cytoskeletal dynamics. These Na+/Ca2+ exchangers are also located in the cell plasma membrane where an influx of Na+ can also cause efflux of H+ ions, resulting in alkalinization of the cytosol and a decrease in perimembrane extracellular pH. This in turn increases the activity of cathepsin proteases to favor ECM degradation and cell invasion, as has been demonstrated in MDA-MB-231 breast cancer cells that overexpress a voltage-gated sodium channel (VGSC).93
Finally, the activity of some sodium channels in prostate and breast cancer cell lines has been shown to stimulate the expression of the same channel, creating a positive feedback loop of channel activity-induced channel expression that the cells can utilize to dramatically increase ion flux.94
VGSCs are the principal sodium ion channel type involved in tumor metastasis, although other channel types such as epithelial sodium channels (ENaCs) can also drive sodium flux in cancer.95 They have a canonical role in generating and propagating action potentials in neurons but are also found in many nonexcitable cells such as glial, endothelial, immune, fibroblasts, osteoblasts, and cancer cell types where they regulate cell fate and homeostasis.96 When expressed in tumor cells, they are predominantly linked to metastatic cells, and while the mechanism is not fully understood, it has been suggested that they modulate the intracellular pH homeostasis to induce cathepsin-dependent invasion.60,93 This channel type can also generate a persistent current to drive Ca2+ influx and influence cell migration via Ca2+-dependent pathways.97 There are nine members of Na+-selective VGSCs that are composed of two types of subunit: 1 alpha (Nav1.1–1.9) and multiple smaller beta (β1–β4), both of which can contribute to metastasis. The alpha subunit forms the Na+-permeable pore, whereas beta subunits are members of the cell adhesion molecule family that modulate the gating of the pore and are also able to function independently of the alpha subunit.98
The three main VGSCs with evidence of metastatic activity, Nav1.5, 1.6, and 1.7, are shown to increase cell motility and invasion in breast, prostate, cervical, and lung cancers. Both Nav1.6 and 1.7 have been found in biopsies from highly invasive cervical cancers and are involved in cervical cancer metastasis.99 Nav1.7 has also been shown to increase cell motility and invasion in human and rat prostate cancer cell lines,100–104 as well as breast,46,50,105 cervical,99 and lung.106 In prostate cancer cells, the expression of Nav1.7 is regulated at the transcriptional level by epidermal growth factor (EGF) and EGF receptor (EGFR) via ERK1/2 pathways.107
Most investigations into the role of VGSCs in metastasis have focused on Nav1.5, particularly in the context of breast cancer. Interestingly, many breast cancers express a neonatal splice isoform of Nav1.5 rather than the adult isoform found in normal tissues. This neonatal isoform is correlated in breast cancer with lymph node metastasis50 and is preferentially expressed by highly metastatic breast cancer cells (MDA-MB-231) over poorly metastatic cell lines (MDA-MB-268) or normal breast epithelial cells (MCF10A).94 It has been suggested that the biophysical properties of neonatal Nav1.5 enable greater Na+ influx than the adult isoform, which may provide a reason why the cells that abnormally express this isoform are metastatic.108 Inhibiting Nav1.5 activity in MDA-MB-231 breast cancer cells by downregulation, knockdown, or pharmacological molecules inhibits cell migration and invasion.50,94,109
While the precise signaling pathway of Nav1.5-mediated metastasis is unclear, two mechanisms have been suggested. First, in colon cancer, Nav1.5 activity and the resulting depolarization of a cell induce the transcription of invasion-related genes such as PKA, Rap1B, MEK, and ERK1/2.110,111 A Na+ current also promotes src family kinase activity and a proinvasive morphology in MDA-MB-231 breast cancer cells.60 Second, Nav1.5 function in breast cancer has been linked to changes in intra- and extracellular pH, in turn activating cathepsins and other proteases to promote ECM degradation and invasion.60,93,112 In MDA-MB-231 breast cancer cells, Nav1.5 changes the pH by colocalizing with the Na+/H+ exchanger (NHE-1) and calveolin-1 in invadopodia of the cell, causing local acidification and subsequent degradation of the ECM by proteases.112 The efflux of Na+ also causes local changes in Vmem, which can be sensed by the cellular actin network. This may modulate F-actin polymerization/depolymerization and invadopodia formation, further promoting a metastatic phenotype.112,113
There is mounting evidence that the β-subunits of Nav1 channels are also independently involved in cancer cell metastasis. They are known to regulate gating and expression of alpha subunits, which can affect the functions described previously.55 Indeed, downregulation of the beta subunits by RNAi in poorly metastatic MCF7 breast cancer cells promotes an invasive phenotype, whereas overexpression in highly metastatic MDA-MB-231 cells decreases invasion.114 Beta subunits are also directly involved in adhesive interactions with plasma membrane integrins, modulating cytoskeleton dynamics and cellular aggregation via trans-homophilic adhesion.98 The β1 subunit and its gene SCN1B are upregulated in invasive lobular breast carcinoma compared with normal breast tissue, and the β1 protein is upregulated in multiple human breast cancer cell lines.114 It is also the predominant beta subunit expressed by MCF7 breast cancer cells, and the mRNA expression level correlates with the amount of Na+ current and cell adhesiveness. However, SCN1B expression is inversely correlated to transwell migration, suggesting that although it modulates Nav1.5 activity there may be separate mechanisms at play for adhesion versus migration.114
Changes in the intracellular Na+ concentration can also alter cellular pH. A change of pH in turn can affect intracellular protein charges, causing a change in protein conformation and thus activity.55 NHE1 is thought to drive the formation of a pH gradient across the membrane in response to regulation by HIF1α in cancer cells early in the transformation process. The resultant pH gradient affects protease-mediated ECM remodeling, such as MMP2, and promotes an invasive phenotype.115,116 As the pH surrounding a tumor decreases, it also can influence cell adhesion via the formation of integrin-mediated focal adhesion contacts.52,115,117 NHE1 is crucial for the polarization of migrating cells by modulating the pH at the outer surface of the cell membrane in lamellipodia.59 Polarization of NHE1 transporters within a cell allows for the development of a gradient of protons increasing from the trailing to the leading edge of lamellipodium in the direction of migration, which then stimulates the assembly/disassembly of focal adhesions and cell migration.69
Potassium
Unlike Ca2+ and Na+, K+ ions predominantly move from the intracellular to extracellular space through their channels, causing cellular hyperpolarization, and are involved in maintaining the steady-state Vmem of a cell. The majority of reports suggest a role of K+ channels in cancer cell proliferation,29,118–120 but there is also evidence that they can regulate metastasis (Fig. 3C). Like Na+, the efflux of K+ indirectly affects Vmem by driving Ca2+ entry into the cell, stimulating the Ca2+-dependent migration pathways. Efflux of K+ can also directly control Ca2+ influx via the Ca2+-activated K+ channel KCa2.3 (SK3), which is found in MDA-MB-435 breast cancer cells as well as neoplastic patient tissue, but not in nontumor breast tissue.61 Cells that express SK3 have a high intracellular Ca2+ concentration, and siRNAs against SK3 can abolish the migration of breast cancer cells.61
Again we see evidence of a positive feedback loop that is co-opted by cancer cells: SK3 is activated by the rising levels of intracellular Ca2+, leading to cell hyperpolarization, which increases the electrochemical driving force for entry of more Ca.2+121 Activation of SK3 in MDA-MB-435 breast cancer cells is coupled to another protein called ionotropic purinergic P2X7 receptor-channel (P2X7R), which is also present in several types of tumors.122 Interaction of SK3 and P2X7 increased cell migration in MDA-MB-435 cells, and P2X7R alone also increased the invasion of cells via an SK3-independent mechanism linked to cathepsin activation.123 SK3 also forms a complex with Orai1 in breast and colon cancer, which may then selectively influence Ca2+ influx.123 A voltage-gated potassium channel, Kv10.1, has also been implicated in Vmem hyperpolarization through Orai1-mediated Ca2+ entry, and interestingly only affects migration and not cell proliferation; Kv10.1 is functionally expressed in MDA-MB-231 breast cancer cells, and silencing the gene results in cell depolarization, reduced Ca2+ influx, and reduced cell migration without change in cell proliferation.123 Another Ca2+-activated potassium channel, KCa3.1 (IK3), was found to be preferentially expressed in several types of metastasizing tumors: breast,124 pancreas,125 and prostate.119 However, the role of KCa3.1 is less clear and appears to be cell-type dependent; in some tumors, expression decreases metastasis whereas in others it leads to an increase.126
There is also some evidence that K+ channels can influence cell adhesion in a Ca2+-independent mechanism. The voltage-gated potassium channels Kv1.3 and Kv11.1 interact with β1 integrin to regulate cell adhesion by K+ activity.52 In addition, the ether-a-go-go potassium channel EAG2 is enriched at the trailing edge of migrating medulloblastoma cells and regulates cell volume dynamics to facilitate cell motility independent of Ca2+ signaling.127 Overall, the effects of K+ on metastasis appear to mainly involve driving Ca2+ flux but may also include Ca2+-independent mechanisms that need to be further explored.
Chloride
The main form of anion transport that accompanies the transport of cations Na+/K+/Ca2+ is chloride. Chloride flux has been predominantly linked to metastasis by way of cell volume regulation, with evidence that Cl− channel activity is required for glioma migration and invasion128–130 (Fig. 3D). To decrease their volume, cells activate K+ and Cl− channels that stimulate KCl and water release. To increase cell volume, Na+/K+/2Cl− cotransporters, Na+/H+ ion exchangers, and cation channels are activated leading to a net uptake of KCl and water. Polarization of these channel types leads to differences in cell volume across a cell to drive migration.126 Driven by K+ and Cl− ion flow, local changes in cell volume facilitate the invasion of tumor cells through constricted interstitial spaces.54
This phenomenon has been characterized most extensively in glioblastoma. Glioblastoma cells express NKCC1, an Na+/K+/Cl− cotransporter, and are thought to use high intracellular Cl− concentrations to decrease their cell volume by driving water out of the cell.131 In addition, local volume increases in lamellipodium can support the outgrowth with concurrent osmotic shrinkage at the trailing edge of the cell. Chloride ion channel-4 (CIC4) is a Cl−/H+ exchanger that has been demonstrated to enhance migration, invasion, and metastasis of both glioma and colon cancer cells by this mechanism.119 To date, the role of Cl− in other aspects of cancer metastasis has not been reported.
Membrane potential
The importance of ion channel activity and the ions themselves has been established, but the third and key component is changes in Vmem that occur as a consequence of ion flux. Due to the thinness of the plasma membrane (5 nm), small changes in Vmem can translate into large EFs, which can control the conformation and thereby activity of voltage-gated ion channels,52 enabling complex positive feedback loops.132Vmem also sets the electrical driving force for Ca2+ influx, that is, hyperpolarization will induce an increase in intracellular Ca2+, influencing several migration pathways. Additional transduction mechanisms, which convert Vmem changes into secondary messenger cascades and changes in transcription in embryogenesis, include the regulation of signaling molecule transporters133,134 and receptor clustering, such as the KRAS family.135
It is not an easy task to decipher the overlapping effects of Vmem, ions, and ion channel activity, as they are inextricably linked bioelectric properties of a cell.53 However, one way in which roles for Vmem have been established in developmental and neoplastic contexts is to show how diverse ion channel functions can lead to the same outcome, as long as the voltage change is the same.136,137 The resting Vmem can fluctuate based on open/close kinetics of ion channels, and ion channel conformation, and thus, activity can change based on Vmem fluctuations, making it unclear how these properties are modulated to facilitate metastasis in cancer cells. However, to effectively target bioelectric signaling pathways for cancer treatment, it is important to understand their relationship.
It is well known that there is considerable heterogeneity in the resting Vmem between, and even within, the same cell type. Proliferative cells such as stem, embryonic, and cancer cells have a more depolarized Vmem than other somatic cells of the body.29,138,139 Traditionally, an excitable cell is defined by the ability to generate an action potential, and while cancer cells cannot generate action potentials, small steady-state inward currents can be generated by Nav1 channels in the window of the cancer cell-resting Vmem.108 This current promotes the depolarization of the cell and an increase in intracellular Na+. The “CELEX hypothesis” proposed by Djamgoz suggests that it is the electrically excitable membrane of metastatic cells, due to the combined effect of inward currents generated by VGSCs and a reduction in voltage-gated potassium channel activity, that promotes metastasis.140 Furthermore, within cancer cells there is variability in the amount of depolarization; a more depolarized Vmem is associated with a higher metastatic potential and forced hyperpolarization of cells can reduce their migration and invasiveness.141–143
Depolarization is thought to have a direct effect on cytoskeletal dynamics through three main pathways. First, direct voltage-dependent activation of ERK, leading to activation of GTP/GDP exchange factor, Rho, then Rho kinase signaling leading to myosin light chain phosphorylation.52,53 Second, depolarization regulates the actin polymerization/depolymerization ratio and thus can control cell stiffness, via direct sensing of transmembrane EFs by the actin network.113 For example, when Vmem hyperpolarizes, hERG1 is activated and interacts with β1 integrins to recruit FAK and Rac1 to the membrane.144,145 Third, Vmem affects the cytoskeleton by controlling the activity of voltage-dependent enzymes. The TRP ion channels TRPM2 and TRPM7 both have domains that interact with enzymes: TRPM2 has adapted the ability to bind ADP-ribose, sensing its accumulation in the cell via activation of Na+/Ca2+ entry through the channel domain,146 whereas TRPM7 contains an Mg2+-regulated protein kinase domain coupled to an Mg2+ channel, and therefore, the kinase activity responds to local changes in free Mg2+, which will be affected by changes in Vmem. Finally, in embryogenesis and regeneration, Vmem can directly regulate gene expression, for example, by modulating the Hedgehog and Notch signaling.147 It is unknown if this regulation can occur in cancer but would be an important aspect of cancer cell Vmem to investigate.
It has been demonstrated that ion flux regulated by ion channel activity and Vmem can mediate metastasis largely through the control of cell migration pathways. Most studies to date focus on a single channel or pathway, however, it is recognized that the flux of one ion influences both other ion types and Vmem, and vice versa; thus, it is not necessarily change in one ion channel that produces a metastatic phenotype. There is also the question of cellular compensation. Do cancer cells have an abnormal Vmem because of dysregulated ion channel activity compared with normal cells, or do they compensate for changes in Vmem by upregulating ion channels or modulating their activity in an effort to re-establish homeostasis?
Evidence shows that cancer cells utilize positive feedback loops to produce bioelectric conditions favorable for metastasis; are cancer cells co-opting strategies used by other cell types for normal functions, or are they simply lacking the regulatory checks and balances of normal cells? For example, neutrophils express proton channels specifically to prevent depolarization of the cell Vmem to counterbalance an influx of Ca2+ produced by other channels for cell migration.148 Many metastatic cells also have an increase in Ca2+ influx but may be lacking in the compensatory mechanisms to prevent depolarization. In future studies, it will be important to look at the summation of these changes to create a dynamic bioelectric profile of the cell.
Extrinsic Bioelectric Properties
A myriad of extrinsic properties of the tumor microenvironment are widely known to regulate the metastatic potential of cancer cells (reviewed elsewhere4). The bioelectric state of a given tissue also represents an important feature of the local microenvironment. Indeed, most organs that are enclosed by a layer of epithelial cells generate an EF known as the transepithelial potential (TEP).149 A biological EF is defined as the electrical potential difference between two points, measured in volts per distance, and is generated by polarized ion transport and current flow.37 Endogenous EFs serve as directional cues during wound healing, embryogenesis, sprouting of endothelial cells, and tumor cell migration.53 The migration of cells in response to an EF is known as electrotaxis.150
Disruption of the skin TEP during wounding causes a shift in local potential, leading to the migration of keratinocytes as part of the healing process.150 As with wounding, there are changes in the TEP with tumor formation. For example, in breast tissue, normal epithelial cells have a TEP gradient of +30 mV generated between the apical and basal sides of the lumen. When a tumor forms in the breast epithelium, some cells will divide more rapidly, causing a break in the symmetry of the TEP and localized membrane depolarization66,151 (Fig. 4, Box 1). This creates an EF early on in tumor progression that is measurable at the surface of the skin.43,44

Cancer cells also sense the trans-endothelial potential of blood vessels, which may be involved in intra/extravasation149,152 (Fig. 4, Box 2). Interestingly, EF signals have been shown to both work synergistically with and even override chemical and other types of microenvironment signals, suggesting that there is a hierarchical arrangement of physical and chemical factors.151 Consequently, it is essential to understand the impact of EF changes that occurs during tumor initiation on local invasion and metastasis and the response to known prometastatic cues.
Local changes in EFs
Cell migration is an essential component of the metastatic activity of a tumor. The application of an in vitro EF of strength comparable with the TEP will elicit electrotaxis in a variety of cancer cell types, and an electrotactic response is positively correlated with metastatic potential.153–155 While exposure to an EF induces polarization of charged species such as sialic acids on the surface of cells, this is not necessary or sufficient to cause motility and an electrotactic response must involve specific changes in cell membrane protein function.156 One of the key early events in normal cell electrotaxis is a polarized increase of intracellular Ca2+; when Ca2+ influx is inhibited, cells can no longer detect and respond to an EF, for example, when VGCCs are blocked with Ni2+ or Sr.2+154,157,158 Polarization of Ca2+ within a cell asymmetrically activates several downstream pathways involved in migration such as receptor tyrosine kinases, PI3K, Rho GTPases, and ERK, which then elicit cytoskeletal changes necessary for directed migration.66–68
The mechanisms known to drive electrotaxis in normal cell types appear to also be involved in electrotactic responses of cancer cells, although the regulation of these mechanisms may differ. Similar to the effects of ion flux, these mechanisms include changes in Ca2+ concentration leading to actin polymerization/depolymerization and cell adhesion, and growth factor signaling (Fig. 4, Box 1). MDA-MB-231 breast cancer and rat mammary adenocarcinoma MTLn3 tumor cells can detect and respond to an EF, undergoing electrotaxis via signaling through the EGFR.66 EGFR mRNA levels correlate with the electrotactic response, and transfection of weakly metastatic tumor cells with human EGFR was demonstrated to lead to enhanced speed and directedness of electrotaxis.66 Also in MDA-MB-231 breast cancer cells, Nakajima et al., demonstrated that the potassium channel Kir4.2 functions to sense EFs by facilitating PIP3 polarization to the leading edge of the migrating cell.159 Finally, lipid rafts have been suggested to play an important role in localizing membrane proteins such as integrins and will polarize when a cell is exposed to an EF, leading to integrin-mediated downstream signaling through RhoA and PI3K.67
Interestingly, sodium channels are also able to drive electrotaxis. Rat prostate and human breast cancer cell line electrotaxis is reduced when tetrodotoxin or other blockers of VGSCs are administered.160,161 Although the mechanism of VGSC-driven electrotaxis is not well understood, it may involve an alteration in Ca2+ concentration, phosphorylation of cytoskeletal components via kinase activation, or direct interaction with the cytoskeleton via the beta subunits as described in previous sections.149
Although many cancer cell types can undergo electrotaxis, their response varies depending both on the type of cancer and the metastatic potential. In vitro, some cancer cell types undergo electrotaxis toward the anode and others to the cathode, however, the mechanism mediating this is unknown.66,160,162 One study investigating electrotaxis in glioblastoma brain tumor-initiating cells observed that they migrate to the anode when cultured on poly-L-ornithine/laminin-coated plastic, and to the cathode when embedded in a 3D hyaluronan/collagen hydrogel.163 This suggests that the physical cues of the ECM may influence the direction of cells. Other factors dictating electrotaxis direction may include the type of ions and/or ion channels that become polarized when exposed to an EF and which mechanism initiates electrotaxis.
Even within the same cell population, there is variation in the mechanism that cells use to respond to an EF. Li et al. tested the migration of H1975 lung adenocarcinoma cells and an isolated subpopulation of cancer stem cell-like cells in the presence of a direct current EF.154 They reported that both cell subtypes exhibited cathodal migration accompanied by a transient intracellular Ca2+ increase that correlated to the metastatic potential of the cells. However, when they probed the mechanism of EF sensing, they found that blocking the activity of stretch-activated cation channels completely inhibited H1975 cell EF-activated intracellular Ca2+ increase, whereas it only partially inhibited Ca2+ increase in the cancer stem cell-like population, suggesting that there are different mechanisms at play in their EF response.
An EF can act not only at the single-cell level but also at the level of a population of cells. Gap junctions allow the immediate transfer of bioelectric signals between cells, setting the membrane potential of individual cells and forming bioelectric networks within groups of cells and on a larger tissue scale within the body. The normal breast epithelium cell line MCF10A was demonstrated to respond differently to an EF in vitro depending on if cells were cultured in isolation or partially confluent.164 Partially confluent or “clustered” cells exhibited electrotaxis with an EF half the strength required to stimulate migration in isolated cells, suggesting that when cells are in contact they are more sensitive to an EF. This effect was shown to be partially dependent on E-cadherin; cell with E-cadherin knockdown exhibited decreased electrotaxis only in the collective group. The study also observed that clustered cells have a higher persistence, likely due to the physical constraints of the neighboring cells, which force a cell to move in the same direction as well as momentum transfer between cells with adhesions.
In light of the ability of bioelectric signals to be propagated by cell/cell contacts, it becomes important to understand how the Vmem of one cancer cell can affect the Vmem of a neighboring cell, or if a noncancer neighboring cell can affect a cancer cell or vice versa. Gap junctions are composed of proteins called connexins and research has shown that there are changes in connexin expression, such as a decreased expression of connexin43 in breast cancer patient samples and cell lines, linked to cancer.165 The data on the role of connexins and metastasis are conflicting; a lack of gap junctions, for example, in MDA-MB-435 breast cancer cells, has been correlated with increased metastasis and invasion,165–167 and also inhibiting gap junctions was reported to lead to reduced migration and invasion in vitro, as well as reduced metastatic burden in the lungs and liver in vivo with MDA-MB-231 breast cancer cells.168
These differences may arise from variability in cell line, specific connexins tested, whether homocellular or heterocellular communication is targeted, or temporal differences in the role of connexins during metastasis, but regardless, presumably a loss of gap junctions between cancer cells would lead to a lack of electrical coupling between cells, a loss of ion exchange, and a loss of the ability to propagate an EF through a cell population. It is unclear how this might fit in with the currently understood relationship between gap junctions and metastasis but is a potential target of future studies.
Long-range propagation of oncogenic signals
Colonization is an important step in metastasis and to achieve this, cells must identify and be able to survive in a suitable secondary environment. Recent work suggests that tumor cells may be able to prime the premetastatic site from a distance before colonization to create a favorable niche, for example, through the secretion of exosomes.169 These observations raise the question of whether bioelectric signaling could be part of this premetastatic niche priming.
One of the remarkable aspects of Burr’s prescient work on the bioelectrics of cancer was that he was able to measure the presence of tumors at a considerable distance.170 Recent molecular work has begun to corroborate this finding with modern tools: bioelectric signals in non-neural tissues propagate long range within the body, carrying information about injury,171 proliferation/apoptosis,172 and regulating the ability of oncogene expression to convert cells into tumors.173,174 This is consistent with a view of cancer as a reversion of cells to an ancient unicellular state, in which the cell views the rest of the body as just an external environment,175,176 and the increasingly apparent role of bioelectric signals in mediating the long-range communication that normally harnesses individual cells into a larger whole—the metazoan organism and its target morphology.23,28,177 Here, we focus on the spatial aspects of bioelectric cues in metastasis specifically.
Work in a tadpole in vivo model revealed that a temporary depolarization, induced by a variety of means (but most conveniently by targeting chloride channels), could convert normal melanocytes to a melanoma-like phenotype (Fig. 4, Box 3). In the absence of any oncogene, mutation, carcinogen, or DNA damage, healthy animals exhibited an extreme hyperpigmentation due to the overproliferation of melanocyte cells, which radically changed the melanocyte morphology and caused invasion into all body regions, including blood vessels, neural tube, brain, and visceral organs.136,178
Two aspects are notable, with respect to common assumptions about cancer. First, there was no primary tumor: every melanocyte in the animal converted. Second, while later these cells expressed markers associated with the EMT (SLUG, etc.), the initial phenotype was driven entirely by physiological change,179 demonstrating how critical signaling in the cancer process can occur via mechanisms that would be undetectable by genomic, transcriptomic, or proteomic profiling.
The mechanism for this effect involves serotonergic signaling: depolarization alters 5HT-mediated signaling, which triggers proliferation and metastatic behavior, and thus can be effectively suppressed with SERT blockers such as fluoxetine.136 Indeed, epidemiological studies have suggested that there is a reduction in colon cancer and glioma risk with selective serotonin reuptake inhibitor (SSRI)180,181 and tricyclic antidepressant182 use, respectively. A key aspect of this is the fact that the signaling is not cell autonomous: the cells in which voltage was changed, to induce change in melanocyte behavior, are not melanocytes themselves, but a different population of cells known as “instructor cells.”136
This sparse but ubiquitous cell population expresses the glycine-gated chloride channel, which provides a convenient way to regulate their bioelectric state specifically. The means by which they are depolarized does not matter, it is the Vmemper se that regulates their ability to trigger conversion in melanocytes via their efflux of serotonin. Remarkably, however, it was found that the depolarization, as well as hyperpolarization that could suppress this process, was not merely noncell autonomous but could occur at very long range.183 A change in Vmem of very few cells at one end of the animal was sufficient to trigger global conversion, including melanocytes at the other end. While the serotonergic mechanism required for this is known, serotonin itself is too small to be fluorescently labeled without changing its transport properties; thus, future advancements in tracking very small signaling molecules in vivo will be necessary to observe the actual propagation of relay signaling across tissue that can trigger distant metastasis. If cancer cells can propagate bioelectric signals to distant sites for metastasis, similarly to what is seen with instructor cells in tadpoles, this raises interesting implications for priming of the premetastatic niche and the mechanism for cancer cell colonization.
Future Outlook
One of the main barriers to the study of bioelectrical signaling in cancer is adapting the tools for measuring and manipulating Vmem and ion channel activity. Many tools were designed for investigation of fast electric changes in excitable cells such as neurons, whereas the changes in nonexcitable cells are often slower and subtler.184 In addition, because bioelectricity is involved in multiple pathways, it can be difficult to establish a clear mechanism for the link between the physical phenomenon of Vmem and biochemical pathways of metastasis. Ion channel gene expression may be a useful biomarker but it does not determine the channel expression or activity, or whether the net effect is due to activity, expression, cross talk between ion channels or other channel types, or a combination of each. Another aspect that makes developing tools to measure bioelectric effects difficult is the temporal fluctuations of Vmem and ion channel activity.
The current gold standard to measure Vmem is patch clamp, but this method is low throughput, unsuitable for in vivo, disruptive to the cell homeostasis, and cannot provide spatial resolution (Lazzari-Dean JR, Gest AMM, Miller EW. Optical determination of absolute membrane potential. Submitted; under review, 2019). The use of voltage-sensitive dyes can be confounded by artifacts due to dye loading or fluorophore bleaching, and hence, there is currently a push to develop more precise optical tools for Vmem visualization. For example, a recent publication demonstrates the use of Voltage-Fluor fluorescence lifetime imaging (VF-FLIM) to record absolute Vmem with single-cell resolution (Lazzari-Dean JR, Gest AMM, Miller EW under review, 2019).
This technology is advantageous because depolarization of Vmem attenuates the rate of photoinduced electron transfer, providing a measure of the direct relationship between fluorescence and Vmem. The researchers demonstrated the utility of VF-FLIM by investigating EGF-induced hyperpolarization of A431 squamous carcinoma cells, finding by blocking K+ currents or intercepting cytosolic Ca2+ and measuring Vmem that it is mediated by Ca2+-activated K+ channel KCa3.1. Continued development and adaptation of the toolkit for cancer cells will help to expand our knowledge of the relationship between bioelectric signaling and metastasis.
The ultimate aim of deciphering bioelectrical control of metastasis is to translate mechanistic studies into clinical strategies. There are two main areas where bioelectric signaling has potential for clinical translation: detection and monitoring of a tumor, and the development of drugs that target the bioelectric state of a tumor cell for treatment. Early detection and consistent monitoring of tumors are an important determinant of patient outcome. Previous work has suggested that it is possible to detect a difference in TEP at the skin surface between malignant and normal tissue in breast cancer43,44 and that bioelectric changes occur before any detectable genetic or molecular changes in the tissue.174
Electrical impedance spectroscopy, which measures TEP through the skin, is inexpensive and easily implemented clinically as it is already used for applications such as cardiac cycle imaging, gastrointestinal function monitoring, and skin cancer detection.185–187 Understanding the significance and role of bioelectric changes at the cellular and tissue level can be used to further the development of this technology to detect changes in the breast tissue using electric potential, both earlier and with greater accuracy than current methods.
The drug development pipeline for cancer treatment is both expensive and lengthy, with up to 1 billion dollars spent per drug and 10 or more years needed for development.188 Due to these hurdles, there has been a shift in focus to the repurposing of existing drugs already approved by regulatory agencies for human use in noncancer treatments.189 Repurposing drugs can reduce the time for drug development as the drug already has well-understood pharmacokinetics in humans and an established safety profile. Many FDA-approved drugs target ion channels, but have not been tested in cancer, creating an opportunity to investigate their repurposing.
The online database Repurposing Drugs in Oncology (ReDO) is a collection of noncancer drugs that have reported evidence of preclinical or clinical anticancer effects.190,191 Of the 280 drugs listed in the ReDO database, 40 affect ion channel function. For example, one potassium channel, Kir4.1, can be blocked via commonly prescribed SSRI, which has been shown to have antitumor effects in colon192 and breast193 cancers.
Serotonin (5-HT) receptors, in particular, have been proposed as a novel target for cancer therapies. The 5-HT3 receptor involves a ligand-gated Na+/K+ ion channel, and 5-HT binding causes membrane depolarization.194,195 The 5-HT3 antagonist Y25130 has a proapoptotic and cell cycle arresting effect on colorectal cancer cells in vitro,196 and the antidepressant drug sertraline was demonstrated to show comparable toxicity with doxorubicin and other chemotherapeutics in colorectal cancer cells in vitro and inhibited tumor growth in vivo.192 Although an effect directed at metastasis is yet to be reported, the wide variety of drugs that target the serotonin pathway are promising candidates for investigation. Drugs that block VGSCs, such as ranolazine for angina and the antiepileptic drug phenytoin, have also been shown to have an in vivo effect on tumor growth and invasion in a mouse model of breast cancer.50,197,198
Understanding how ion channels contribute to tumor progression and metastasis may inform new treatment options using repurposed drugs that can target these channels. The large number of ion channel drugs already approved for human use provides cancer researchers with a powerful toolkit of electroceuticals that can be used, together with simulation platforms, to design therapeutic cocktails.199 Identification of already approved drugs will allow faster implementation into the clinic for patients with metastatic disease.
One emerging set of tools that will benefit the study of the role of bioelectricity in cancer is in the area of machine learning. It is clear now that cancer is a very complex system-level disease. Thus, it is very likely that soon (if not already) the known signaling pathways will outstrip the ability of human scientists to mentally compose biorealistic models that have a predictive value. Thus, the complexity of the disease, together with the exponentially increasing literature on this topic, requires that we develop machine learning tools to assist human scientists, not merely in analysis of “big data” but also in the formulation of hypotheses and models.200,201 Crucially in this field, it must involve not only genomic data but also physiological data and the results of functional experiments. Parallel efforts in developmental biology are ongoing toward a next-generation “bioinformatics of shape,”202 of which developments should also be very useful for the cancer field.
Initial efforts in this direction have been made in the context of bioelectrically mediated conversion of melanocytes. It had been observed that various treatments produce a melanoma phenotype with different rates of efficiency (penetrance); however, it was always an all-or-none effect at the level of the animal: any given tadpole converts or does not, with some frequency in the population. The process was stochastic, but the random decision was made coherently by all the melanocytes within each animal.136,179 Seeking to understand this process, a machine learning platform was created, which evolved a model, based on all of the known pathway information relevant to this signaling pathway179: the AI-derived model was able to not only reproduce the data from all past functional experiments but could also be interrogated to suggest interventions and predict outcomes.
It was specifically analyzed to suggest a treatment that would break the concordance among melanocyte decision-making—something that was not previously achieved. The system analyzed this evolved network’s state space and made a hypothesis about what process enabled melanocytes to make decisions; it suggested a complex intervention—two drugs and a protein misexpression, which when tested at the bench, produced the first discordant tadpoles containing both normal and converted melanocytes.202 While this work remains to be fully spatialized and integrated with known long-range bioelectric pathways, it represents a roadmap for using machine learning to discover human-understandable models of complex system-level events, make quantitative predictions, and suggest candidate therapeutics that can be tested in vivo.
Conclusions
There is an urgent need to both understand the mechanism of metastasis and develop new treatment strategies. Bioelectric signaling in cancer and particularly metastasis is a growing field of study, which has demonstrated importance in many of the key mechanisms of metastasis. Most of the data to date focus on the processes of cell migration and invasion, highlighting a lack of insight into the role of bioelectricity in other stages of metastasis such as intra/extravasation and colonization.


